

Exercise 3. Importing, aligning and building trees
(adapted from Geneious Tutorials)
The aim of this exercise is to become familiar with importing DNA nucleotide sequences, aligning them and then analysing the output.
You should in the end produce a phylogenetic tree based on cytochrome b of the following species of the Ursidae family:
GIANT PANDA (Ailuropoda melanoleuca)
MALAYAN SUN BEAR (Helarctos malayanus)
SLOTH BEAR (Melursus ursinus)
ASIATIC BLACK BEAR (Selenarctos thibetanus)
SPECTACLED BEAR (Tremarctos ornatus)
BLACK BEAR (Ursus americanus)
POLAR BEAR (Thalarctos maritimus)
BROWN BEAR (Ursus arctos)
-
A.No Geneious
Porque o alinhamento de todas as sequências de citocromo b de Ursidae demora muito tempo, mesmo quando se usam programas em servidores externos, temos de efectuar uma selecção das 300 e muitas sequências.
-
1.Crie uma pasta nova (File/New Folder);
-
2.Na secção NCBI, Nucleotide faça a busca pretendida;
-
3.Depois da busca completa, seleccione todas as sequências e arraste-as para a pasta recém-criada.
-
4.Faça a selecção das sequências eliminando numa primeira fase:
-
5.genomas completos (mitochondrial complete genomes), cujo tamanho é de cerca de 16000-17000 bases.
-
6.sequências do tipo PREDICTED (records predicted by automated computational analysis, derived from a genomic sequence annotated using gene prediction method)
-
7.sequências de citocromo b incompleto (sabem do exercício anterior o número de bases do gene completo, pelo que tudo o que tiver menor comprimento deve ser eliminado).
-
8.Depois desta eliminação deverá ter um pouco mais de 100 sequências, sendo pois necessário proceder à eliminação de sequências até obter uma lista que não ultrapasse as 30 sequências.
-
9.Que critérios utilizar? Sendo o objectivo a construção da filogenia da família, devem ser retidas sequências que representem todas as espécies e sub-espécies da família abaixo listadas. No entanto, e devido à referida quebra de monofilia dos ursos pardos, e para recuperar um filogenia correcta, deve ter sequências de ambos os clades representados. Assim escolha 2 sequências de cada um dos seguintes grupos: UAU18870-74 e EU567100-110.
-
B.The sequences are not yet aligned despite the fact that all the sequences are from the same mitochondrial gene. You can do the alignment using whatever defaults settings you see fit. Take into consideration the type of nucleotide sequences you have. Inspect the alignment visually.
C. Phylogenetic reconstructions
As you scan the alignment you have constructed you should notice that the DNA sequences are very similar, but not identical. If you look closer at some of the nucleotide differences you will probably be able to see that some bear sequences are more closely related than others. Rather than eyeball the sequences to guess evolutionary relationships it is possible to use the DNA changes to statistically infer the evolutionary relationships from DNA sequences. This modelling is known as phylogenetics. In this exercise we will build a phylogeny of the Ursidae family (bears). Select the alignment that you generated and click on the tree icon in the menu. If you decide to use Geneious you will be prompted for a number of options. These options alter the way that the program models DNA sequences on a tree - in this example we are building a very simple tree. Under Genetic Distance Model select: HKY and UPGMA for the tree building method. Leave the other boxes unchecked then click OK.
Once Geneious has finished constructing the tree select the graphical tree view tab. This is a phylogenetic reconstruction of the bear alignment you made. A few things you should know about phylogenetic trees:
1) The tips correspond to extant (not extinct!) taxa
2) A tree summarises the relatednes of all taxa (i.e a family tree)
3) The internal nodes, or internal vertices correspond to ancestral (hypothetical) taxa
4) The branch lengths represent evolutionary distances. Longer branches mean less sequence similarity. For relatively short branches (say below 0.2) branch length is roughly 1 - the percentage of differing sites in the two sequences. i.e. a distance of .0.04 means 96% sequence similarity.
From the phylogenetic tree you have constructed answer the following questions:
C.1. Which two bear species in your phylogenetic tree are most closely related?
C.2. Which bear species is the most basal on the phylogenetic tree?
C.3. In Geneious when "Show Branch Labels" is checked the length of each branch is shown above it. Use this information to compute the genetic distance of S. thibetanus and U. americanus back to their common ancestral node. What is approximately the % of sequence divergence?
C.4. Compare the number above with actual data. Select the two sequence in the viewer and look at the statistics panel. What is the observed % sequence divergence? Is there a difference?
C.5. If this mitochondrial gene mutated at 1% per million years (an approximate rate for mitochondrial DNA) then how many years ago did the common ancestor of S. thibetanus and U. americanus diverge?
C.6. Given that S. thibetanus has an Asian/Russian distribution and and U. americanus an American distribution what geographical "feature" may have caused the speciation "event" in the common ancestor of these bears.
D. You have just constructed a tree of all the extant (living) bears there are also two bears that have gone extinct in the past 20,000 years. There is a Genbank record for the mitochondrial cytochrome B gene of the extinct cave bear (Ursus spelaeus). This DNA sequence was isolated from a fossil bone - the retrieval of "old" degraded DNA is known as ancient DNA. It is technically challenging, as the DNA is degraded into small pieces (typically 100-200 base-pairs). The goal of this exercise is to find the cytochrome B DNA sequences for Ursus spelaeus and find its closest living relative by integrating it into your existing bears phylogenetic tree.
Go to the NCBI/Nucleotide search and input: Ursus spelaeus cytochrome B and search. By doing this you are searching GenBank for a species and gene name - which is often the easiest way to locate DNA sequences. You can pick up the correct DNA entry from this search window and import it in the appropriate folder. Check that the file now appears in this folder.
In Geneious one of the columns to the right of the sequence summary window is labelled "name". At present the Genbank number AF264047.1 has been inputted. Click on this name and change it to Ursus spelaeus. This will ensure that the species name appears in your tree.
Now select the alignment you generated earlier at the same time as the new Ursus spelaeus sequence you have just imported (hold down contrl key and click to select multiple items). Then click the alignment button and perform the alignment (as done previously). Once alignment is complete build a new tree (as before). View the new tree.
D.1. What are the closest living relative(s) of the extinct cave bear?
D.2. What differences are there between your tree and the tree below?
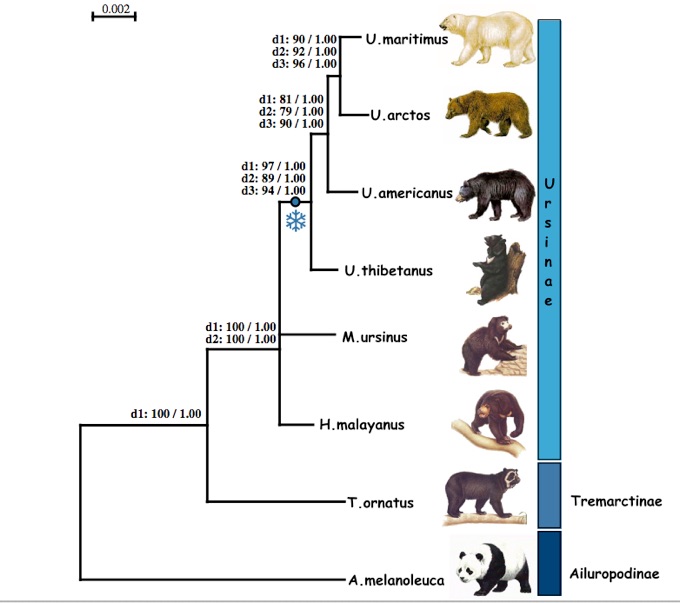
Pagès, M., Calvignac, S., Klein, C., Paris, M., Hughes, S., & Hänni, C. (2008). Combined analysis of fourteen nuclear genes refines the Ursidae phylogeny. Molecular Phylogenetics and Evolution, 47(1), 73-83. doi:10.1016/j.ympev.2007.10.019
